Link iNaturalist observations to TRY
Link iNaturalist vascular plant observations to the previously created trait TRY summary statistics.
This section covers:
Load data
Link iNat and TRY
Fuzzy merge
Log trait values
Number of observations per trait
Plot observation density after linking
Packages¶
import pandas as pd
import os
import numpy as np
# fuzzy matching
#import rapidfuzz
from rapidfuzz import process, fuzz
# plotting
import matplotlib.pyplot as plt
import seaborn as sns
from matplotlib.colors import LogNorm, Normalize
import cartopy.crs as ccrs # plot maps
from matplotlib.colors import BoundaryNorm
from matplotlib.ticker import MaxNLocator
from mpl_toolkits.axes_grid1 import make_axes_locatableLoad data¶
We load the iNaturalist vascular plant observations and the TRY summary stats per species.
iNat = pd.read_csv("Data/iNat/observations.csv")
iNat.head(3)
Load trait measurments with consolidated species name:
TRY = pd.read_csv("Data/TRY/TRY_summary_stats.csv")
TRY.head(2)TRY.shape(51908, 19)iNat.shape(14019405, 6)# check that we have only unique observation ID's
iNat["gbifID"].nunique()14019405Link iNaturalist and TRY¶
Non-fuzzy merge with TRY summary stats on consolidated TRY species name:
iNat_TRY = pd.merge(iNat, TRY,
left_on= ['scientificName'],
right_on= ['AccSpeciesName'],
how='inner')
iNat_TRY.head(3)We repeat the same with the ‘original’ species name in TRY:
TRY_syn = pd.read_csv("Data/TRY/TRY_summary_stats_syn.csv")
TRY_syn.head(2)Extract from TRY those observations that have not been matched:
# filter for observations not in merged dataframe:
iNat_rest = iNat[~iNat.gbifID.isin(iNat_TRY['gbifID'])]
iNat_rest.shape(2541013, 6)# non-fuzzy merge with TRY summary stats on original TRY species name:
iNat_TRY_syn = pd.merge(iNat_rest, TRY_syn,
left_on= ['scientificName'],
right_on= ['SpeciesName'],
how='inner')
iNat_TRY_syn.head(3)subsets = [iNat_TRY, iNat_TRY_syn]
iNat_TRY_all = pd.concat(subsets)iNat_TRY_all = iNat_TRY_all.drop(['AccSpeciesName', 'SpeciesName'], axis = 1)iNat_TRY_all.head(3)iNat_TRY_all.shape(11806220, 24)iNat_TRY_all["gbifID"].nunique()11806220# agian filter for observations not in merged dataframe:
iNat_rest_2 = iNat[~iNat.gbifID.isin(iNat_TRY_all['gbifID'])]
iNat_rest_2.shape(2213185, 6)Check how much was matched:
print('iNat species:')
print(iNat["scientificName"].nunique())
print('TRY consolidated species:')
print(TRY["AccSpeciesName"].nunique())
print('TRY original species:')
print(TRY_syn["SpeciesName"].nunique())
print('species merged:')
print(iNat_TRY_all["scientificName"].nunique())
print('iNat species not merged:')
print(iNat_rest_2["scientificName"].nunique())
# percentage of iNat observations linked with at least one TRY trait
print('percentage of iNat observations linked with at least one TRY trait:')
print(len(iNat_TRY_all)/len(iNat))iNat species:
90820
TRY consolidated species:
51908
TRY original species:
61180
species merged:
27783
iNat species not merged:
63037
percentage of iNat observations linked with at least one TRY trait:
0.8421341704587321
Fuzzy merge¶
Get only unique species names left in iNaturalist unmatched observations:
iNat_rest_unique = iNat_rest_2.drop_duplicates(subset=['scientificName'])Get only unique unmatched TRY species names:
pd.options.mode.chained_assignment = None
TRY = pd.read_csv("Data/TRY/TRY_summary_stats.csv")
TRY_alt = pd.read_csv("Data/TRY/TRY_summary_stats_syn.csv")
TRY_rest = TRY[~TRY.AccSpeciesName.isin(iNat_TRY_all['scientificName'])]
TRY_alt_rest = TRY_alt[~TRY_alt.SpeciesName.isin(iNat_TRY_all['scientificName'])]
TRY_alt_rest.rename(columns = {'SpeciesName':'AccSpeciesName'}, inplace = True)
TRY_R = pd.concat([TRY_rest, TRY_alt_rest])
TRY_rest_unique = TRY_R.drop_duplicates(subset=['AccSpeciesName'])
TRY_rest_unique.head()# define choices and queries
# this might take a little while
choices = TRY_rest_unique["AccSpeciesName"].apply(str)
queries = iNat_rest_unique["scientificName"]
score_sort = [(x,) + i
for x in queries
for i in process.extract(x, choices, score_cutoff=90, scorer=fuzz.token_sort_ratio) ]
fuzzy_matches = pd.DataFrame(score_sort)
fuzzy_matches.head()Save fuzzy match to .csv:
fuzzy_matches.to_csv("Data/fuzzy_matches.csv", sep = "\t",index=False)Reload fuzzy matches:
fuzzy_matches = pd.read_csv("Data/fuzzy_matches.csv", sep = "\t")
fuzzy_matches.head()Add new names to unmatched iNaturalist observations: iNat_rest_2 with fuzzy_matches
fuzzy_matches.rename(columns = {'0':'scientificName'}, inplace = True)
fuzzy_matches.rename(columns = {'1':'fuzzyName'}, inplace = True)
iNat_rest_fuzzy = pd.merge(iNat_rest_2, fuzzy_matches, on='scientificName', how='inner')Merge with TRY:
TRY = pd.read_csv("Data/TRY/TRY_summary_stats.csv")
TRY_alt = pd.read_csv("Data/TRY/TRY_summary_stats_syn.csv")
TRY.rename(columns = {'AccSpeciesName':'fuzzyName'}, inplace = True)
iNat_TRY_fuzzy_1 = pd.merge(iNat_rest_fuzzy, TRY, on='fuzzyName', how='inner')
iNat_TRY_fuzzy_rest = iNat_rest_fuzzy[~iNat_rest_fuzzy.gbifID.isin(iNat_TRY_fuzzy_1['gbifID'])]
iNat_TRY_fuzzy_1= iNat_TRY_fuzzy_1.drop(columns=["fuzzyName", "2", "3"])
TRY_alt.rename(columns = {'SpeciesName':'fuzzyName'}, inplace = True)
iNat_TRY_fuzzy_2 = pd.merge(iNat_TRY_fuzzy_rest, TRY_alt, on='fuzzyName', how='inner')
iNat_TRY_fuzzy_2= iNat_TRY_fuzzy_2.drop(columns=["fuzzyName", "2", "3"])# merge fuzzy-consolidated species name match and fuzzy-original match
frames = [iNat_TRY_fuzzy_1, iNat_TRY_fuzzy_2]
iNat_TRY_fuzzy_merge = pd.concat(frames)iNat_TRY_fuzzy_merge['gbifID'].nunique()/len(iNat_TRY_fuzzy_merge['gbifID'])0.9933762301286904Drop iNat observation duplicates in fuzzy matches, keeping the row with the least NaN
iNat_TRY_fuzzy_merge_2 = (iNat_TRY_fuzzy_merge.assign(counts=iNat_TRY_fuzzy_merge.count(axis=1))
.sort_values(['gbifID', 'counts'])
.drop_duplicates('gbifID', keep='last')
.drop('counts', axis=1))iNat_TRY_fuzzy_merge.shape(89828, 24)iNat_TRY_fuzzy_merge_2.shape(89233, 24)Concatenate to make final dataframe:
frames = [iNat_TRY_all, iNat_TRY_fuzzy_merge_2]
iNat_TRY_final = pd.concat(frames)Compare shape to number of unique gbif ID’s, check that they are the same. We want each observation represented only once:
iNat_TRY_final.shape(11895453, 24)iNat_TRY_final['gbifID'].nunique()11895453iNat_TRY_final.head()After matching with alternate name and a conservative fuzzy match, we were able to match about 85% of the iNaturalist observations with trait information. Many rare species seem to be absent in either one of the two databases.
print('percentage of iNat observations linked with at least one TRY trait:')
print(len(iNat_TRY_final)/len(iNat))
print('percentage of species in iNaturalist matched with TRY:')
print(iNat_TRY_final["scientificName"].nunique()/iNat["scientificName"].nunique())
print('percentage of species in TRY matched with iNaturalist:')
print(iNat_TRY_final["scientificName"].nunique()/TRY["fuzzyName"].nunique())percentage of iNat observations linked with at least one TRY trait:
0.8484991338790769
percentage of species in iNaturalist matched with TRY:
0.3161528297731777
percentage of species in TRY matched with iNaturalist:
0.5531517299838176
iNat_TRY_final.to_csv("Data/iNat_TRY.csv", index=False)Log trait values¶
The cwm in sPlot were caluclated after being log e transformed, so we must log e transform iNat data also:
trait = iNat_TRY_final.columns[6:24]
iNat_TRY_final.loc[:, trait] = np.log(iNat_TRY_final[trait])iNat_TRY_final = iNat_TRY_final.replace(-np.inf, np.nan)
iNat_TRY_final = iNat_TRY_final.replace(np.inf, np.nan)iNat_TRY_final.to_csv("Data/iNat_TRY_log.csv", index=False)Number of observations per trait¶
iNat_TRY_final.head()iNat_TRY.count().round(decimals=-5)gbifID 11500000
scientificName 11500000
decimalLatitude 11500000
decimalLongitude 11500000
eventDate 11500000
dateIdentified 11400000
AccSpeciesName 11500000
Dispersal unit length 4700000
Leaf Area 4800000
SLA 7600000
Leaf C 5000000
LDMC 6700000
Leaf fresh mass 2700000
Leaf N per area 5700000
Leaf N per mass 7000000
Leaf delta15N 2400000
Leaf N P ratio 3800000
Leaf P 5100000
Plant Height 9500000
Seed mass 10200000
Seed length 3500000
Seeds per rep. unit 4000000
Stem conduit density 1200000
SSD 3400000
Conduit element length 300000
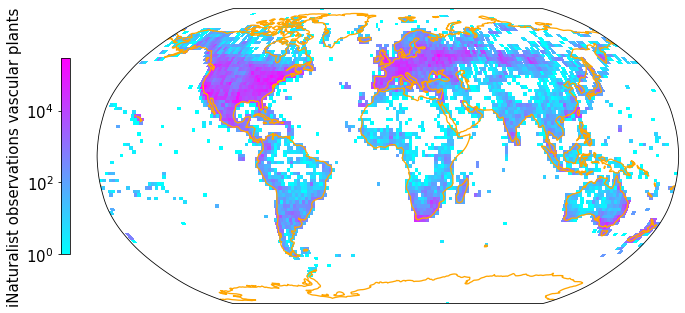
dtype: int64Density of observations after linking¶
plt.rcParams.update({'font.size': 15})
Z, xedges, yedges = np.histogram2d(np.array(iNat_TRY['decimalLongitude'],dtype=float),
np.array(iNat_TRY['decimalLatitude']),bins = [181, 91])
data_crs = ccrs.PlateCarree()
#for colorbar
cmap = plt.get_cmap('cool')
im_ratio = Z.shape[0]/Z.shape[1]
#plot map
fig = plt.figure(figsize=(12, 12)) # I created a new figure and set up its size
#create base plot of a world map
ax = fig.add_subplot(1, 1, 1, projection=ccrs.Robinson()) # I used the PlateCarree projection from cartopy
ax.set_global()
#add coastlines
ax.coastlines(resolution='110m', color='orange', linewidth=1.3)
#add grid with values
im = ax.pcolormesh(xedges, yedges, Z.T, cmap="cool", norm=LogNorm(), transform=data_crs)
#add color bar
#divider = make_axes_locatable(ax)
#cax = divider.append_axes("right", size="3%", pad=0.05)
#fig.colorbar(im, cax=cax)
fig.colorbar(im,fraction=0.046*im_ratio, pad=0.04, shrink=0.3, location="left", label="iNaturalist observations vascular plants")
plt.savefig('Figures/iNat_density_Robinson_TRY.pdf', bbox_inches='tight')